上一篇10x单细胞数据分析我们介绍了如何用Seurat对单细胞数据进行分群分析,这一篇我们介绍一下多个单细胞样品的分析方法。
测试数据选择Seurat提供的ifnb数据集,其中包含两个PBMC样品,一个为干扰素刺激处理,另一个为对照。打开Rstudio,在控制台中输入
devtools::install_github(‘satijalab/seurat-data’)
library(Seurat)
library(SeuratData)
library(patchwork)
InstallData(“ifnb”)
LoadData(“ifnb”)
下载好的ifnb数据是一个Seurat对象,我们需要按照处理方式把它拆成两个样品的对象
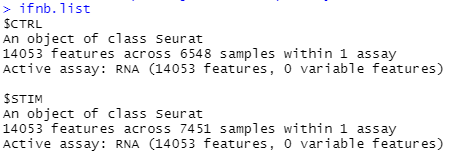
ifnb.list <- SplitObject(ifnb, split.by = “stim”)
输入ifnb.list查看拆分后的对象
Seurat在进行多个单细胞样品分析时,有两种处理方式,直接合并和批次效应校正后合并。我们分别介绍这两种分析方法。
1、直接合并
直接使用merge函数将两个样品合并,这个方法直接将两个对象内的数据矩阵分别合并到一起,生成一个新的Seurat对象
merged.ifnb<- merge(x = ifnb.list[[1]],y = ifnb.list[2])
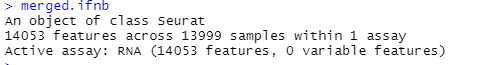
合并后的数据可以当成一个单样品的Seurat对象进行分析
merged.ifnb <- NormalizeData(merged.ifnb, normalization.method = “LogNormalize”, scale.factor = 10000)
merged.ifnb <- FindVariableFeatures(merged.ifnb, selection.method = “vst”, nfeatures = 2000)
merged.ifnb <- ScaleData(merged.ifnb)
merged.ifnb <- RunPCA(merged.ifnb)
merged.ifnb <- FindNeighbors(merged.ifnb, dims = 1:30)
merged.ifnb <- FindClusters(merged.ifnb)
merged.ifnb <- RunUMAP(merged.ifnb, dims = 1:30)
用DimPlot查看合并对象的分群结果
p1 <- DimPlot(merged.ifnb, reduction = “umap”, group.by = “stim”)
p2 <- DimPlot(merged.ifnb, reduction = “umap”, label = TRUE, repel = TRUE)
p1 + p2

左边的为两个样品的细胞在UMAP结果中的分布,右边的为细胞cluster在UMAP中的分布。可以看到两个样品几乎没有重叠的细胞群。
2、批次效应矫正
Seurat提供了一组Integration方法来去除批次效应,这些方法首先识别处于匹配生物学状态(anchors)的跨数据集细胞对,然后基于这些anchors校正数据集之间的批次效应

首先提取用来进行Integration的基因,然后找到anchors,基于anchors进行批次效应矫正
ifnb.list <- lapply(X = ifnb.list, FUN = function(x) {
x <- NormalizeData(x)
x <- FindVariableFeatures(x, selection.method = “vst”, nfeatures = 2000)
})
features <- SelectIntegrationFeatures(object.list = ifnb.list)
ifnb.anchors <- FindIntegrationAnchors(object.list = ifnb.list, anchor.features = features)
ifnb.combined <- IntegrateData(anchorset = ifnb.anchors)
ifnb.combined是两个样品经过批次效应矫正后合并的Seurat对象,对这个对象进行分群分析
DefaultAssay(ifnb.combined) <- “integrated”
ifnb.combined <- ScaleData(ifnb.combined, verbose = FALSE)
ifnb.combined <- RunPCA(ifnb.combined, npcs = 30, verbose = FALSE)
ifnb.combined <- RunUMAP(ifnb.combined, reduction = “pca”, dims = 1:30)
ifnb.combined <- FindNeighbors(ifnb.combined, reduction = “pca”, dims = 1:30)
ifnb.combined <- FindClusters(ifnb.combined)
用DimPlot查看合并对象的分群结果

可以看到经过批次矫正后,两个样品的大部分细胞在UMAP上都是重叠的。
使用Seurat提供的细胞类型信息对两种分析的分群结果进行注释。

测试样品经过批次矫正后,相同类型的细胞都聚到了一起,这样更有利于对细胞类型进行鉴定。实际分析时,我们可以对自己的样品同时做这两种分析,根据结果来判断是否需要进行批次矫正。
3、差异分析
可以对相同细胞类型在不同的样品间的表达差异进行分析,这里以B细胞为例。
首先,从对象中提取出B细胞
DefaultAssay(ifnb.combined) <- “RNA”
B.ifnb <- subset(ifnb.combined, idents = “B”)
将B细胞按照样品类型分开
Idents(B.ifnb) <- “stim”
使用FindMarkers比较两个样品间的差异
b.interferon.response <- FindMarkers(B.ifnb, ident.1 = “STIM”, ident.2 = “CTRL”, verbose = FALSE)

可以用VlnPlot可视化一下差异基因在不同细胞类型中的表达
VlnPlot(ifnb.combined, features = c(“LYZ”, “ISG15”, “CXCL10”), split.by = “stim”, group.by = “celltype”, pt.size = 0,ncol=1)

获得的差异基因还可以进一步的挖掘,如GO/KEGG富集分析等。这里就不展开了,后续的单细胞分析再一一分析吧!