甲基化分析pipeline开发步骤详解
已完成的甲基化Gitee项目地址
如果你只想获得一个现成的pipeline工具加到你的linux服务器上,可以直接到我的Gitee项目地址clone下去,并跳过下面的具体代码问题解决过程。
项目代码仓库:
甲基化项目Gitee地址
.
甲基化背景知识
甲基化是一种常见的基因修饰,研究人员对样本进行特定实验处理后,希望观测到实验样本和对照样本的差异甲基化情况。
生物信息角度关于甲基化通俗的来说就是获得的测序文件如fastq文件中甲基化的碱基会被测序为C,未甲基化的C碱基会被测序为T,所以甲基化分析前期可以理解为字符串处理问题,后期为统计学问题。
甲基化详细解释链接:
甲基化知乎详解
.
开发准备工作
你需要一个好的编辑器,这里不推荐vim,notepad等,对于复杂的项目来说,最好还是使用VS studio或者VS code,因为随时可以添加自己需要的插件如Git等。
-
本教程使用的程序语言:
python,R; -
本教程使用的代码编辑器:
VS code ;
功能模块拆解
我们预期会将pipeline分解成如下几个模块
-
预处理模块:
读取配置文件为变量,生成中间结果文件夹和最终结果文件夹; -
Bismark主流程模块:
fastq文件质量控制,bismark比对去重,从bam提取甲基化信息生成包含甲基化位点cov文件 ; -
样本的甲基化位点PCA投影:
以第二步cov文件为输入,组合成矩阵,进行pca降维并绘制投影图; -
差异甲基化位点calling:
以第二步cov文件为输入,使用DSS软件,进行wald检验得到差异甲基化位点; -
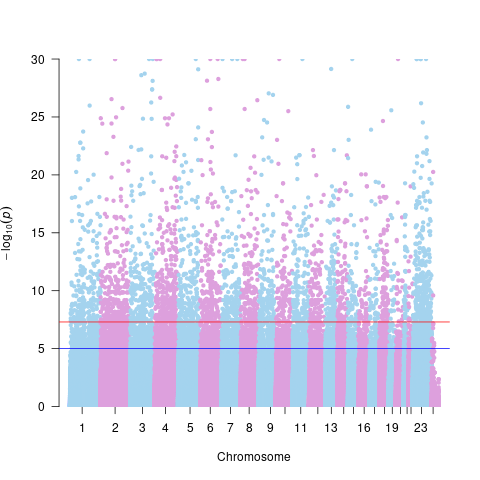
差异甲基化位点的Manhattan图:
以第二步cov文件为输入,组合成矩阵,进行pca降维并绘制投影图; -
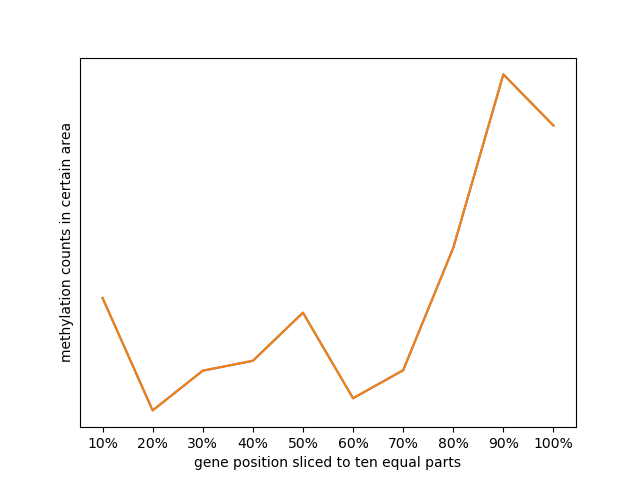
基因十等分统计甲基化位点比例图:
以第二步cov文件为输入,按照甲基化位点在染色体上的位置,统计每个染色体区间上的位点比例并绘制折线图; -
甲基化基因的volcano图:
用差异甲基化位点得到的甲基化程度和pvalue绘制volcano图; -
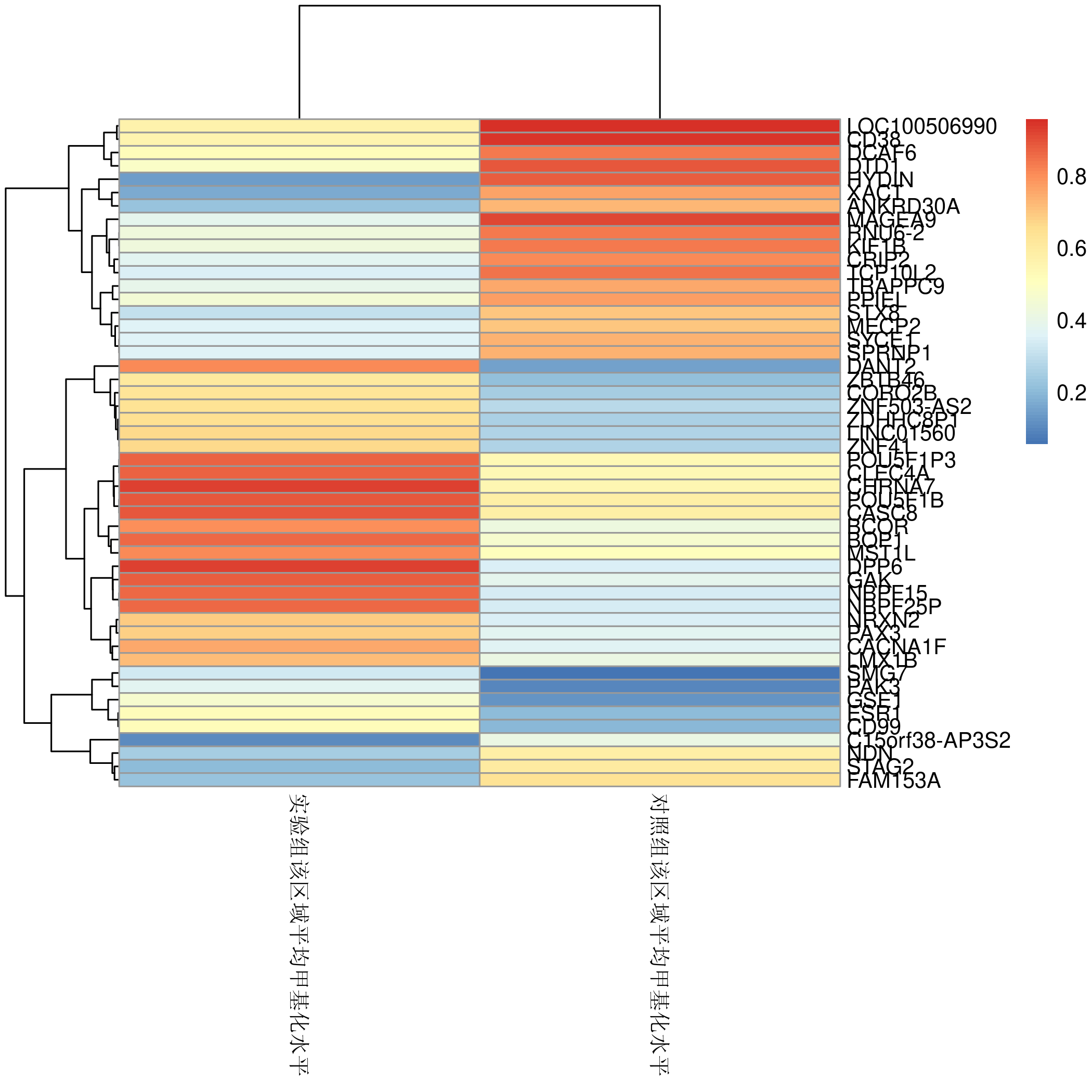
甲基化基因的heatmap图:
用差异甲基化位点得到的甲基化程度绘制heatmap图; -
pipeline运行结束后发送状态码到API:
本步骤如果存储运行状态,失败或者成功状态到数据库,对于web架构比较重要,如果项目无前端,本步骤可忽略;
项目详细解决方案
1.
预处理模块:
本模块只要为了获取config.json的内容并存储为变量,并生成一些文件夹存放运行结果。
config.json为pipeline配置文件,存储样本信息,分组信息,结果路径等。
{"sampth_path":[["/home/DUAN/methyl_rawdata/yinle/J-H-0_1.fq.gz","/home/DUAN/methyl_rawdata/yinle/J-H-0_2.fq.gz"],["/home/DUAN/methyl_rawdata/yinle/J-H-5_1.fq.gz","/home/DUAN/methyl_rawdata/yinle/J-H-5_2.fq.gz"],["/home/DUAN/methyl_rawdata/yinle/293FT_1.fq.gz","/home/DUAN/methyl_rawdata/yinle/293FT_2.fq.gz"],["/home/DUAN/methyl_rawdata/yinle/293FT-HOSK_1.fq.gz","/home/DUAN/methyl_rawdata/yinle/293FT-HOSK_2.fq.gz"],["/home/DUAN/methyl_rawdata/yinle/J-H-10_1.fq.gz","/home/DUAN/methyl_rawdata/yinle/J-H-10_2.fq.gz"]],"control_name":["J-H-0","J-H-5"],"treat_name":["293FT","293FT-HOSK","J-H-10"],"result_path":"/public/mlrna_seq/sample_result/26/","id":"26"}
项目没有采用command line传参的模式,因为参数过多而且字符串长度较大,所以用配置文件的方式。
下面用python库json读取config.json的内容并存储为变量供后续运行使用。
import json
with open("/public/mlrna_seq/make_txt/config.json",'r') as load_f:
load_dict = json.load(load_f) #读取json文件为python字典形式
files = load_dict["sampth_path"] #第一个命令行变量,样本名称,用,分隔,如sample1,sample2
control_name = load_dict["control_name"]
treat_name = load_dict["treat_name"]
group = ",".join(control_name + treat_name)
index = load_dict["result_path"] #结果路径
reid = load_dict["id"]#任务id
用python库os在linux上生成文件夹,加上-p参数可以防止报错。
os.system("mkdir -p "+index)
path="/home/DUAN/methylation/"
os.system("mkdir -p "+path+"analysis")
os.system("mkdir -p "+path+"analysis/pre_data")
2.
Bismark主流程模块:
这里需要安装软件并加入linux path,建议使用conda安装,需要安装bismark和fastp软件,同时从GitHub下载bowtie2源文件放到任意文件夹,bismark需要建立索引操作,bismark详细使用教程链接:
bismark使用方法
,如下两个命令轻松解决。
conda install -c bioconda bismark
conda install -c bioconda fastp
下面代码说细节就是用fastp质控一下fastq文件中的碱基,然后用bismark比对去重,最后用extract_CpG_data.py从bam文件中提取甲基化位点生成cov文件。
cov = []
for fil in files:
result = fil
fil = result[0].split("/")[-1]
os.system("mkdir -p "+path+"analysis/result/"+fil) #为每个样本名在result路径下生成一个文件夹
if len(result) == 1: #结束不符合要求的单端测序
print("请确保是双端测序")
exit(0)
else:
file1 = path+"analysis/pre_data/"+fil+"_R2_pre"
file2 = path+"analysis/result/"+fil+"/"+fil+"_R1_pre_bismark_bt2_pe.sam"
file3 = path+"analysis/result/"+fil+"/"+fil+"_R1_pre_bismark_bt2_pe.deduplicated.sam"
file4 = path+"analysis/result/"+fil+".cov"
if not Path(file1).exists(): #开始fastp流程
os.system("fastp -w 8 -i "+result[0]+" -o "+path+"analysis/pre_data/"+fil+"_R1_pre "+"-I "+result[1]+" -O "+path+"analysis/pre_data/"+fil+"_R2_pre -h "+path+fil+".html")
os.system("cp "+path+fil+".html "+index)
if not Path(file2).exists(): #开始bismark比对流程
os.system("bismark --temp_dir /home/DUAN --bowtie2 -p 4 --path_to_bowtie /public/DUAN/methylation/bismark_ref/bowtie2-2.3.4.2-linux-x86_64 -N 0 -L 20 --quiet --un --ambiguous --sam -o "+path+"analysis/result/"+fil+" /home/lvhongyi/lvhy/reference/human/ensembl_hg19/bismark_ref -1 "+path+"analysis/pre_data/"+fil+"_R1_pre"+" -2 "+path+"analysis/pre_data/"+fil+"_R2_pre")
os.system("echo "+fil+" >> "+index+"mapping.txt") #开始将比对结果写入文件,包含覆盖度信息
os.system("grep -w Mapping "+path+"analysis/result/"+fil+"/*_PE_report.txt >> "+index+"mapping.txt")
if not Path(file3).exists(): #开始bismark去重流程
os.system("deduplicate_bismark -p "+path+"analysis/result/"+fil+"/"+fil+"_R1_pre_bismark_bt2_pe.sam"+" --output_dir "+path+"analysis/result/"+fil)
if not Path(file4).exists(): #开始从bismark结果提取甲基化信息生成cov文件流程
os.system("python extract_CpG_data.py -i "+path+"analysis/result/"+fil+"/"+fil+"_R1_pre_bismark_bt2_pe.deduplicated.sam"+ " -o "+path+"analysis/result/"+fil+".cov")
cov.append(fil+".cov")
上述流程比较简单,用if语句判断目标文件是否存在,防止二次运行。
3.
样本的甲基化位点PCA投影:
下面的代码涉及一种编程思想,动态生成变量。采用动态生成变量的原因就是事先不知道变量的数量,所以无法预先定义。实际总变量数量通常与循环次数有关。这里采用python globals() 函数,可以把globals理解为包含变量的字典。
count = 1
names = globals()
all_set = set()
pca_file = index+group+".pca.txt"
if len(cov)!=2:
if not Path(pca_file).exists():
for covs in cov: #
names['n' + str(count)] = {}
names['a' + str(count)] = set()
with open(path+"analysis/result/"+covs) as f:
for i in f:
pos = i.split("\t")[0]+i.split("\t")[1]
globals()['a' + str(count)].add(pos) #给set赋值,找寻公共染色体位置
globals()['n' + str(count)][pos] = int(i.split("\t")[3]) / int(i.split("\t")[2]) #按照染色体位置存储每个样本的单位点甲基化值
if count>=2:
all_set &= globals()['a' + str(count)]
else:
all_set = globals()['a' + str(count)]
count+=1
dataframe_dic = {}
for sett in all_set:
dataframe_dic[sett] = [globals()['n' + str(t)][sett] for t in range(1,count)]
df = pd.DataFrame(dataframe_dic,index=[i.split("/")[-1].split(".")[0] for i in cov])
df = df.iloc[:,np.random.randint(1,df.shape[-1],20000)] #只选取了20000个位点
df.to_csv(pca_file)
os.system("Rscript /public/DUAN/methylation/pca.R "+pca_file+" "+index+"pca.png")
上面问题的核心就是找到样本A里和样本B里都有的基因位点。
4.
差异甲基化位点calling:
该步骤为本项目核心,通过统计学建模去寻找实验组和对照组的差异甲基化位点。
本步骤可选择的工具较多,涉及的统计学思想也很多。可以应用别人开发好的,也可以自行开发算法,关键是提出假设,并对假设进行验证。下面给出两种解决办法,请读者根据实际需求,自行决定。
使用已经开发好的R package DSS:
先安装好R依赖
BiocManager::install("DSS")
DSS详细教程
教程地址
.
DSS核心是将甲基化reads看作离散变量,建立beta-binomial统计学模型,最后用wald检验理论和真实值的差异,从而找到差异位点。
library(DSS)
library(bsseq)
Args <- commandArgs()
numOfArgs<-length(commandArgs())
file_names<- unlist(strsplit(Args[6],split=",")) #获取文件名字
bseq_names<- unlist(strsplit(Args[7],split=",")) #获取bseq obj命名
bseq_names<-as.vector(bseq_names)
group1<- as.vector(unlist(strsplit(Args[8],split=",")))
group2<- as.vector(unlist(strsplit(Args[9],split=",")))
result_file<- Args[10] #获取结果文件名字信息
result_file1<- Args[11] #获取结果文件名字信息
list_names<-''
lis<-list()
for(i in 1:length(file_names)){ #生成dat1,dat2变量存储甲基化cov信息
name<-paste("dat",as.character(i),sep = "")
if(list_names==''){
list_names<-name
}
else{
list_names<-paste(list_names,name,sep = ",")
}
assign(name,read.table(file_names[i], header = T,col.names=c("chr","pos","N","X")))
lis[[i]]<-get(paste("dat",as.character(i),sep = ""))
}
BSobj <- makeBSseqData(lis,bseq_names) #主分析流程
dmlResult <- DMLtest(BSobj, group1 = group1, group2 =group2, smoothing = T)
dmls = callDML(dmlResult,p.threshold = 1) #,p.threshold = 0.001,delta = 0.1
write.table(dmls,sep="\t",row.names =FALSE,file=result_file1)
dmrs = callDMR(dmlResult ,p.threshold = 0.05,delta = 0.1)
write.table(dmrs,sep="\t",row.names =FALSE,file=result_file)
DSS需要特定的参数,样本名,分组信息正确才能准备分析。
自行开发算法:
同时我也自行开发了一个算法,这里的假设是甲基化测序得到的reads无法反映实际甲基化程度,那么这个理论值就可以通过建模得到,这里建立一个hidden Markov模型,将甲基化描述为三种状态:未甲基化,部分甲基化,完全甲基化,从第一个甲基化CpG位点出发,下一个位点有三种可能性,每种transition state的概率可以视为1/3或者通过观测值统计求出,建模得到的理论值和观测值通过fisher或者卡方检验即可得到差异位点。
展示部分分析结果
Manhattan图
heatmap图
十等分切割基因位置统计图
问题反馈与协助
项目代码仓库:
甲基化项目Gitee地址
.
有任何问题可以到代码仓库提问。