一、数据准备
10X单细胞转录组理论上有3个文件才能被读入R进行seurat分析,分别是barcodes.tsv 、 genes.tsv和matrix.mtx,文件barcodes.tsv 和 genes.tsv,就是表达矩阵的行名和列名
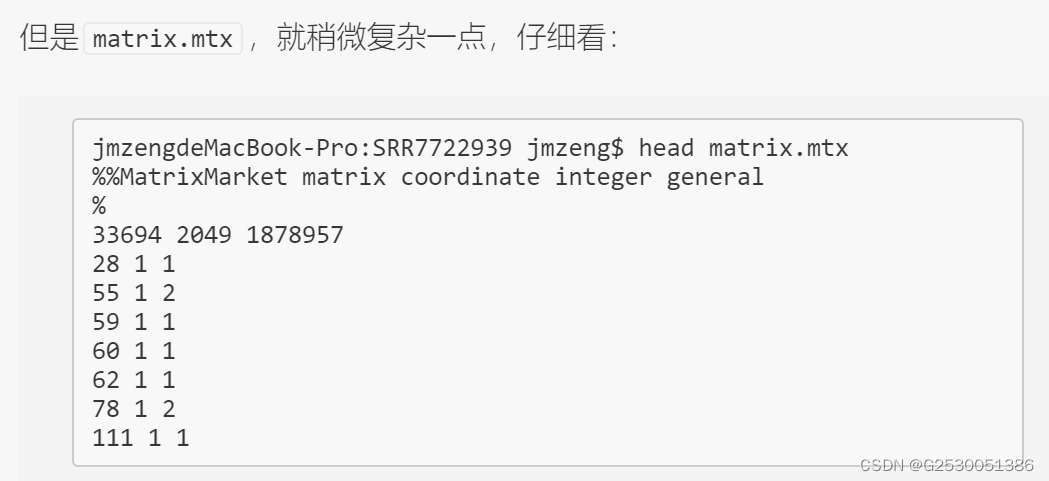
就会发现,matrix.mtx文件里面的33694、2049、1878957数值,分别是细胞数量,基因数量,以及有表达量的值的数量。
下游处理的时候,一定要保证这3个文件同时存在,而且在同一个文件夹下面,每一个样本都是3个文件,每一个样本都是同样的代码处理。
二、一般流程
(一)数据前处理:质控和数据过滤
1.基于QC度量的细胞选择与筛选(即质控)
2.数据标化与缩放(即数据标准化)
3.高度可变特征的检测(特征性基因的选择)
(二)PCA分析:线性降维
PCA分析,并且找到后续数据处理的
维度
(三)细胞聚类
将具有相似基因表达模式的细胞之间绘制边缘,然后将他们划分为一个内联群体
并进行tSNE和UMAP分析
(五)差异分析:寻找marker gene
通过差异表达找到每个聚类的marker gene,差异分析可以有多种形式,如找到所有聚类的marker gene(如cluster1中所有的markgene是指cluster1相对于其余所有cluster是差异的)、两个cluster之间的差异分析、某个cluster中两个样品之间差异分析等
(六)可视化标记基因,即细胞注释
三、
质控分析(QC)
1
.
为什么要做质控?
在细胞分离过程中的细胞损伤或者文库制备的失败(无效的逆转录或者PCR扩增失败),往往会引入一些低质量的数据。这些低质量的数据的主要特点是:
细胞整体上的counts值少(列)
基因的低表达(行)
线粒体基因或者spike-in的比例相对较高
如果这些损伤的行或者列,没有被移除的话,可能会对下游的分析结果产生影响。所以我们在进行分析之前,一定要率先移除这些低质量的行与列。(一开始的理解,后面整个流程做完之后,或许理解会更多,那么接下来在做详细的补充)
2、质控的指标
每一个细胞所有基因的counts值之和
在文库制备的过程中,可能因为细胞的裂解或cDNA捕获和扩招效率的低下,而使得RNA的丢失。
具有较小的counts值之和的细胞被认为是低质量的细胞,考虑被去除。
每一个细胞中单个基因的表达数量
多样化的转录本如果没有被成功的捕获到,因此任何一个细胞中有很少的基因表达,被认为是低质量的,考虑被去除。
每一个细胞中,spike-in序列/线粒体基因占总的counts值的比例
每个细胞中添加的spike-in序列(
人为添加的表达量的参照系
)的浓度都是等量的。
如果spike-in的比值很高,那么就意味着在实验的过程中,大量的转录本丢失。
四、PCA分析
PCA(Principal Component Analysis),主成分分析,是一种使用最广泛的数据降维算法。PCA的主要思想是将n维特征映射到k维上,在尽可能保留原始数据信息的同时降低数据维度来加速数据分析。
过程就是从原始高维的空间中按顺序地找一组相互正交的坐标轴系统,新的坐标轴的选择与数据本身是密切相关的。
其中,第一个新坐标轴选择是原始数据中方差最大的方向,第二个新坐标轴选取是与第一个坐标轴正交的平面中使得方差最大的,第三个轴是与第1,2个轴正交的平面中方差最大的。依次类推,可以得到n个这样的坐标轴。大部分方差都包含在前面k个坐标轴中,后面的坐标轴所含的方差几乎为0。于是,我们可以忽略余下的坐标轴,只保留前面k个含有绝大部分方差的坐标轴,实现对数据降维。
五、确定数据集的维度
目的:每个维度(pc)本质上代表一个“元特征”,它将相关特征集中的信息组合在一起。因此,越在顶部的主成分越可能代表数据集。然而,我们应该选择多少个主成分才认为我们选择的数据包含了绝大部分的原始数据信息呢?
方法:(1)JackStraw()函数, 使用基于零分布的置换检验方法。随机抽取一部分基因(默认1%)然后进行pca分析得到pca分数,将这部分基因的pca分数与先前计算的pca分数进行比较得到显著性p-Value。根据主成分(pc)所包含基因的p-value进行判断选择主成分。最终的结果是每个基因与每个主成分的关联的p-Value。保留下来的主成分是那些富集小的p-Value基因的主成分。JackStrawPlot()函数提供可视化方法,用于比较每一个主成分的p-value的分布,虚线是均匀分布;显著的主成分富集有小p-Value基因,实线位于虚线左上方。
(2)“ElbowPlot函数,基于每个主成分所解释的方差百分比的排序,通过寻找“拐点”来判断几个维度可包含数据的大部分信息。