该R包在全基因组测序WGS中可以通过用于描述突变位点在染色体上的分布,在转录组测序RNA-Seq中可用于描述差异表达基因在染色体上的分布,在WGBS中可用于描述DNA甲基化在染色体上的分布等。
R包软件安装
以下代码在RStudio中执行
install.packages("RIdeogram")
require(RIdeogram)
载入测试数据
data(human_karyotype, package="RIdeogram")
data(gene_density, package="RIdeogram")
data(Random_RNAs_500, package="RIdeogram")
# 查看人类核型数据
head(human_karyotype)
#> Chr Start End CE_start CE_end
#> 1 1 0 248956422 122026459 124932724
#> 2 2 0 242193529 92188145 94090557
#> 3 3 0 198295559 90772458 93655574
#> 4 4 0 190214555 49712061 51743951
#> 5 5 0 181538259 46485900 50059807
#> 6 6 0 170805979 58553888 59829934
# 查看基因密度数据
head(gene_density)
#> Chr Start End Value
#> 1 1 1 1000000 65
#> 2 1 1000001 2000000 76
#> 3 1 2000001 3000000 35
#> 4 1 3000001 4000000 30
#> 5 1 4000001 5000000 10
#> 6 1 5000001 6000000 10
# 查看标签信息数据
head(Random_RNAs_500)
#> Type Shape Chr Start End color
#> 1 tRNA circle 6 69204486 69204568 6a3d9a
#> 2 rRNA box 3 68882967 68883091 33a02c
#> 3 rRNA box 5 55777469 55777587 33a02c
#> 4 rRNA box 21 25202207 25202315 33a02c
#> 5 miRNA triangle 1 86357632 86357687 ff7f00
#> 6 miRNA triangle 11 74399237 74399333 ff7f00
对于自己的数据集可使用下列方法载入:
human_karyotype <- read.table("karyotype.txt", sep = "\t", header = T, stringsAsFactors = F)
gene_density <- read.table("gene_density .txt", sep = "\t", header = T, stringsAsFactors = F)
Random_RNAs_500 <- read.table("Random_RNAs_500 .txt", sep = "\t", header = T, stringsAsFactors = F)
下载基因注释gff文件
GENCODE FTP下载链接 ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_32/gencode.v32.annotation.gff3.gz

GFFex函数提取热图信息
gene_density <- GFFex(input = "gencode.v32.annotation.gff3.gz", karyotype = "human_karyotype.txt", feature = "gene", window = 1000000)
全基因组数据绘制人类染色体密度图
ideogram(karyotype = human_karyotype, overlaid = gene_density)
convertSVG("chromosome.svg", device = "png")
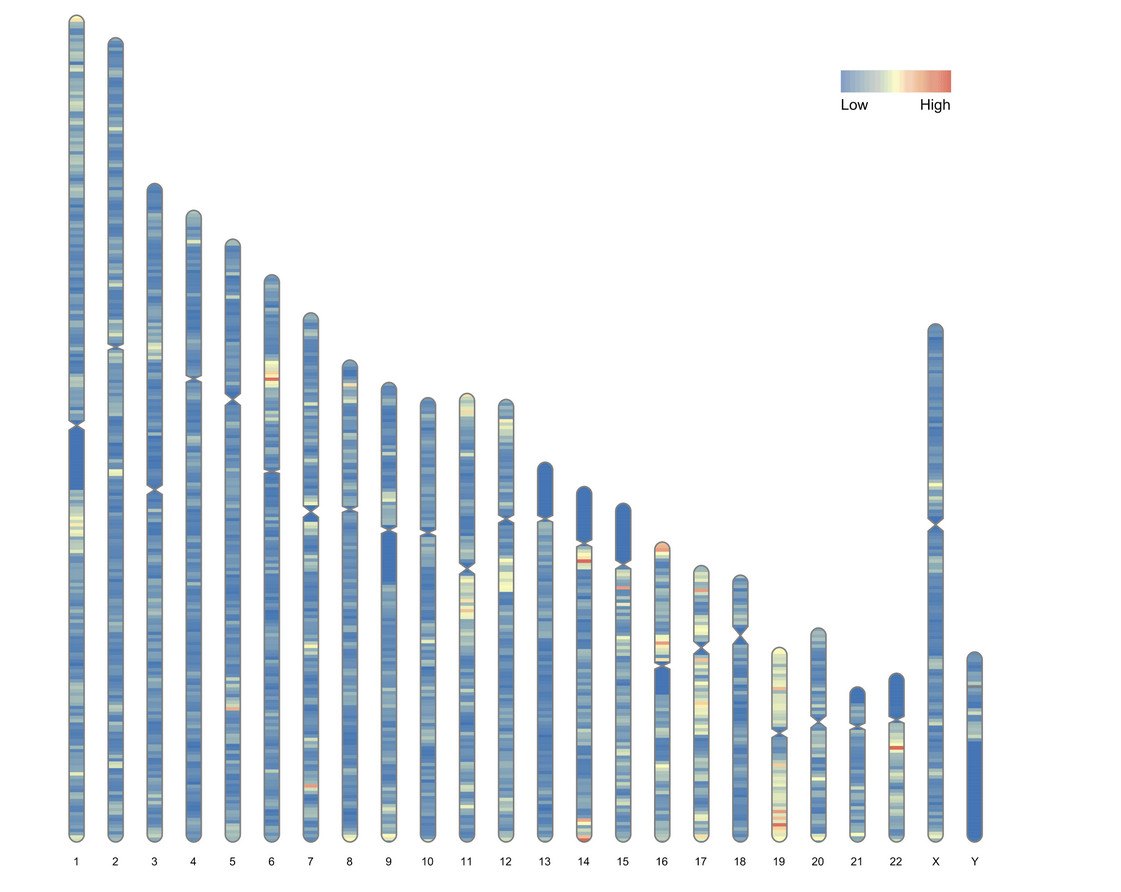
绘制带有标签的染色体密度图
对于无着丝数据信息,可使用human_karyotype[,1:3]取核型数据的前三列。
ideogram(karyotype = human_karyotype[,1:3], overlaid = gene_density, label = Random_RNAs_500, label_type = "marker")
convertSVG("chromosome.svg", device = "png")
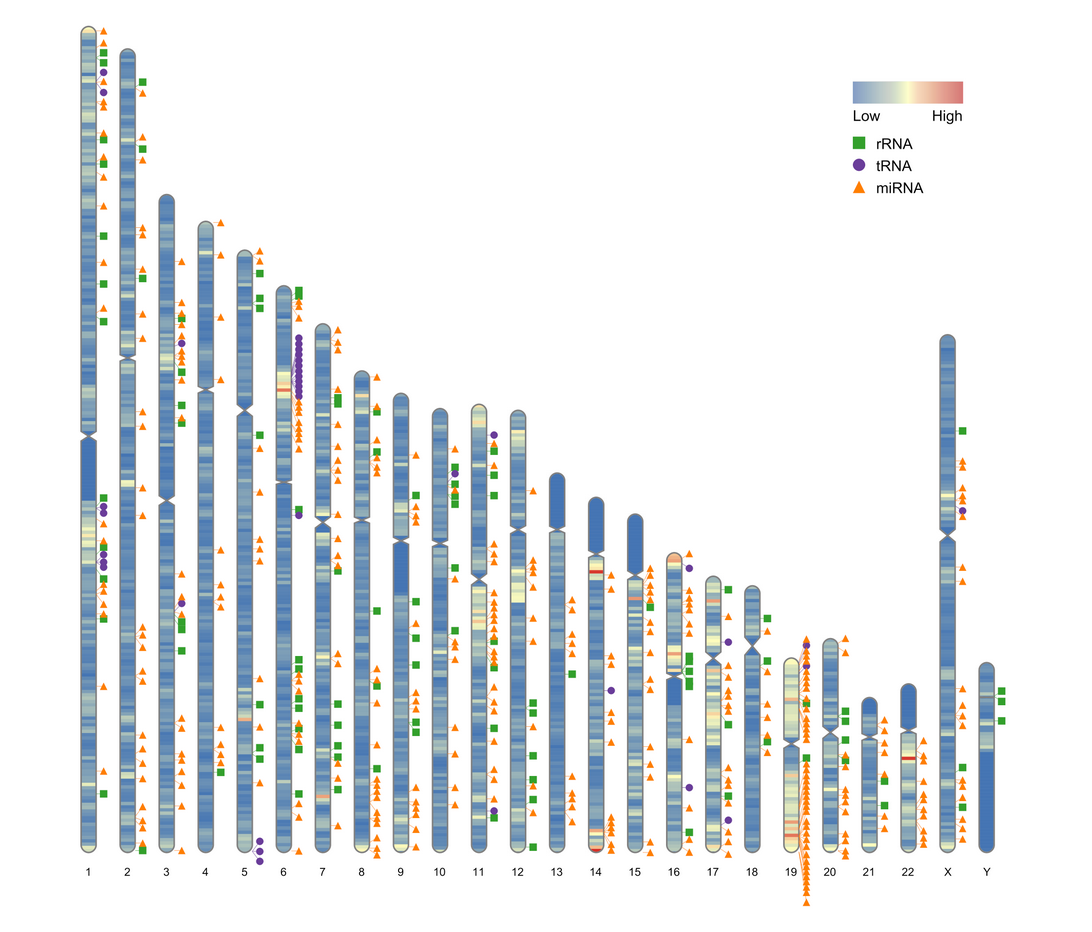
如果只想绘制部分染色体的密度图,例如1号-10号染色体,可使用以下代码:
human_karyotype <- human_karyotype[1:10,]
ideogram(karyotype = human_karyotype, overlaid = gene_density, label = Random_RNAs_500, label_type = "marker")
convertSVG("chromosome.svg", device = "png")
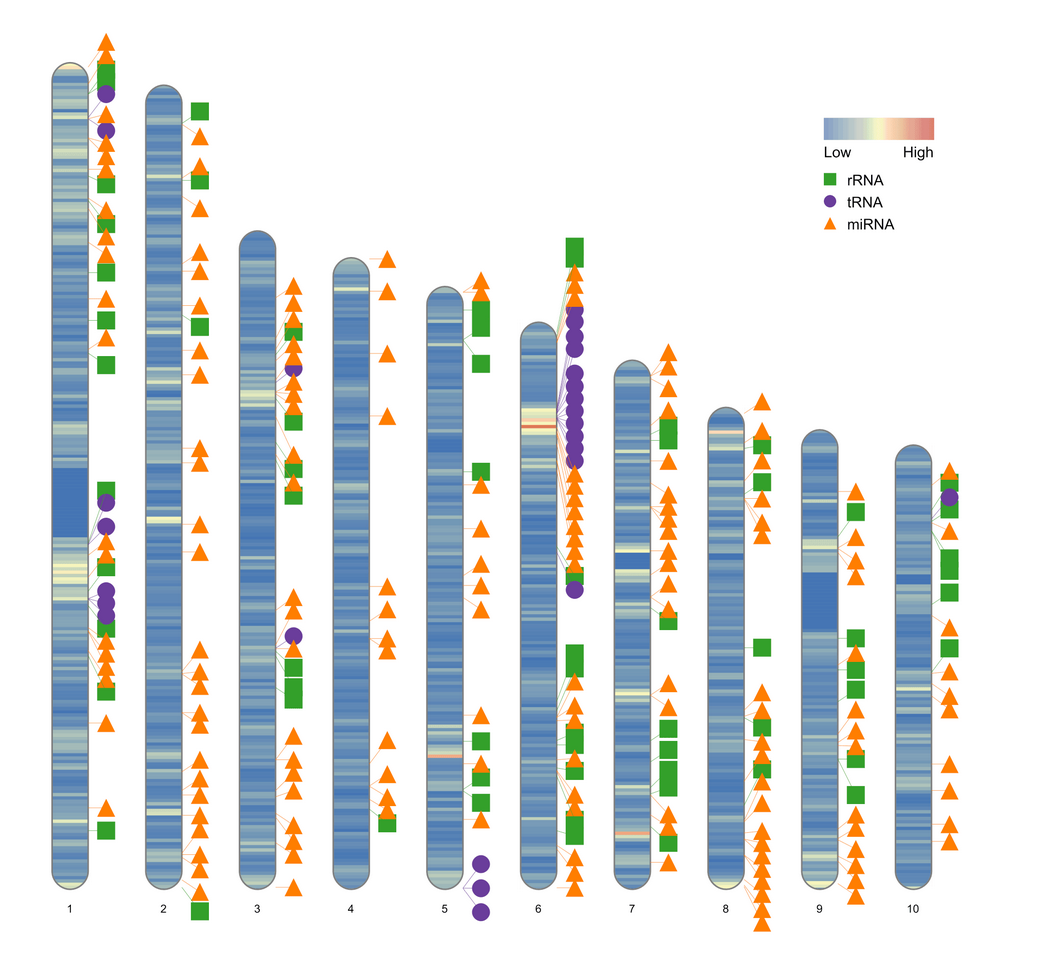
绘制一对染色体的密度图
data(human_karyotype, package="RIdeogram") #reload the karyotype data
ideogram(karyotype = human_karyotype, overlaid = gene_density, label = LTR_density, label_type = "heatmap", colorset1 = c("#f7f7f7", "#e34a33"), colorset2 = c("#f7f7f7", "#2c7fb8")) #use the arguments 'colorset1' and 'colorset2' to set the colors for gene and LTR heatmaps, separately.
convertSVG("chromosome.svg", device = "png")
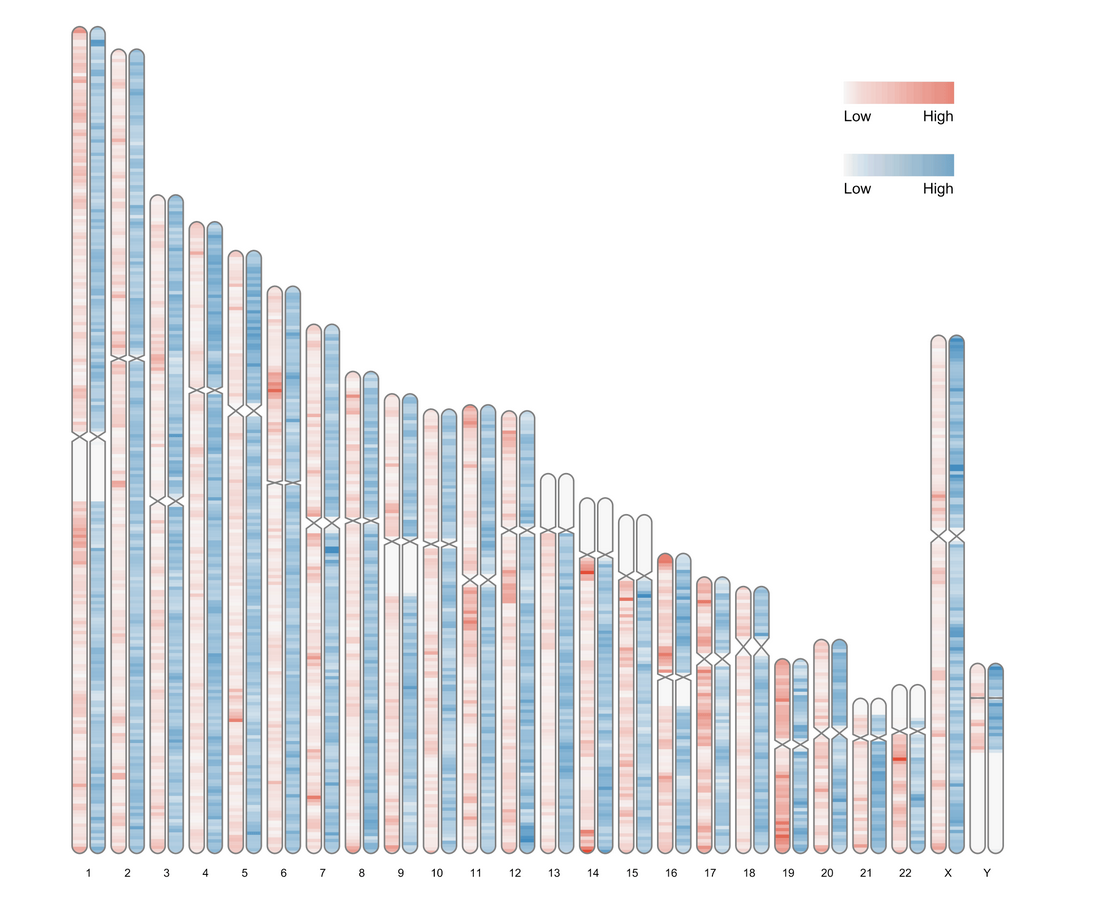
图片导出设置
支持导出tiff, pdf, jpg,png图像格式,也可以设置dpi值(默认为300)
convertSVG("chromosome.svg", device = "tiff", dpi = 600)
# 将svg转换为其他格式文件
svg2tiff("chromosome.svg")
svg2pdf("chromosome.svg")
svg2jpg("chromosome.svg")
svg2png("chromosome.svg")
生信软件1 – 测序下机文件比对结果可视化工具 visNano
版权声明:本文为LittleComputerRobot原创文章,遵循 CC 4.0 BY-SA 版权协议,转载请附上原文出处链接和本声明。