DESeq2包分析需两个矩阵:
1 count矩阵(必须为matrix)
行为Sample id
2 group矩阵(matrix或dataframe)
有Sample和Group的对应关系,
顺序不重要
;
Group列
变量类型为factor;
library("TCGAbiolinks")
library("stringr")
library("DESeq2")
一、读入数据
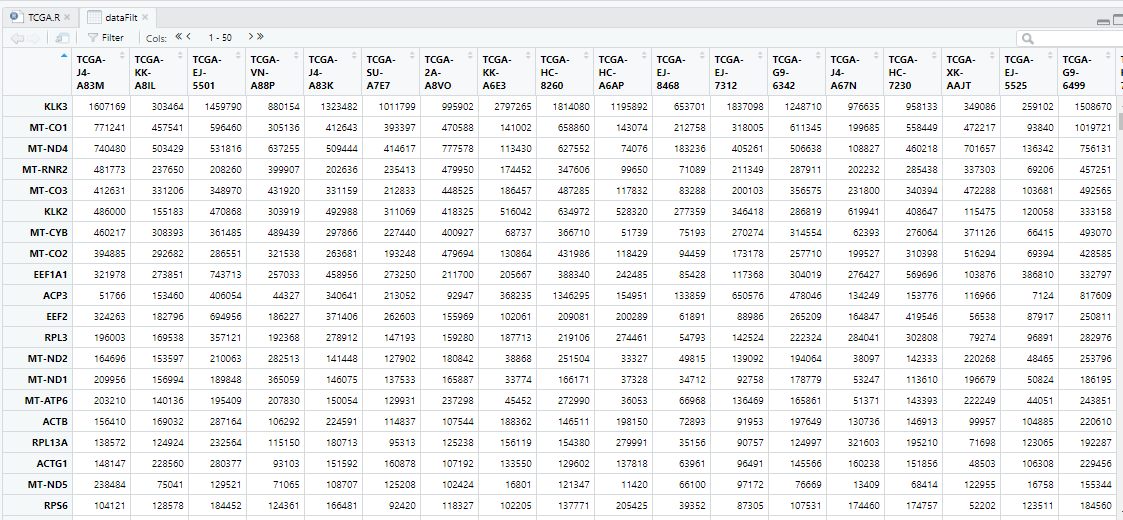
dataFilt=read.csv(file = "Results/测序or临床数据下载/dataFilt.csv", header=T, row.names=1,check.names=FALSE)
dataFilt=as.matrix(dataFilt)
colnames(dataFilt)=str_sub(colnames(dataFilt),1,12) #取病人编号,即前12位
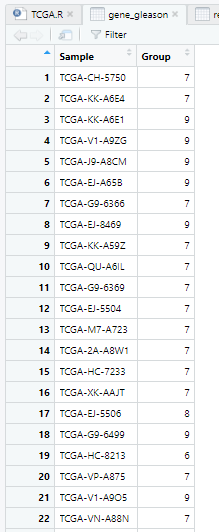
gene_gleason=read.csv(file = "Results/不同gleason评分的SMC4表达/gene_gleason.csv", header=T, row.names=1,check.names=FALSE)
gene_gleason=gene_gleason[,-2]
colnames(gene_gleason)[2]='Group'
View(dataFilt)
View(gene_gleason)
二、确定分组
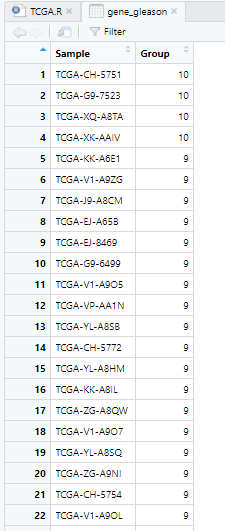
1 gleason评分降序排列
t_index=order(gene_gleason$Group,decreasing = T)
gene_gleason=gene_gleason[t_index,] #调整Sample顺序,使gleason评分降序排列
rownames(gene_gleason)=c(1:nrow(gene_gleason)) #行名从1开始
View(gene_gleason)
2 分组,gleason大于等于8为high_gleason,小于等于7为low_gleason
gene_gleason$Group[1:197]='high_gleason'
gene_gleason$Group[198:nrow(gene_gleason)]='low_gleason' #制定分组
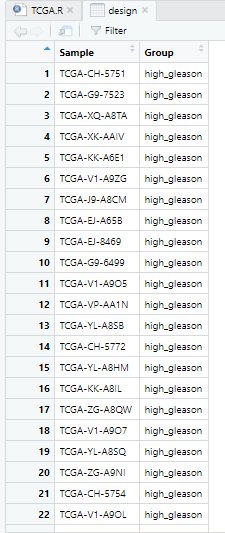
3 得到
group矩阵
,即design
design=gene_gleason
design$Group=as.factor(design$Group) #character转factor
View(design)
三、DESeq2分析
上述的
dataFilt、design
即为构建好的
count矩阵、group矩阵
1 dds、dds1、res三步走
dds <- DESeqDataSetFromMatrix(countData = dataFilt, colData = design, design= ~Group) #countData需要一个count matrix,colData需要一个有分组信息的dataframe,design需要指定分组信息中的列
dds1 <- DESeq(dds,fitType = 'mean')
res <- results(dds1, contrast = c('Group', 'high_gleason', 'low_gleason')) #Group对应上面的“design= ~Group”,且high_gleason在前,low_gleason在后
2 输出结果
result <- data.frame(res, stringsAsFactors = FALSE, check.names = FALSE)
View(result)
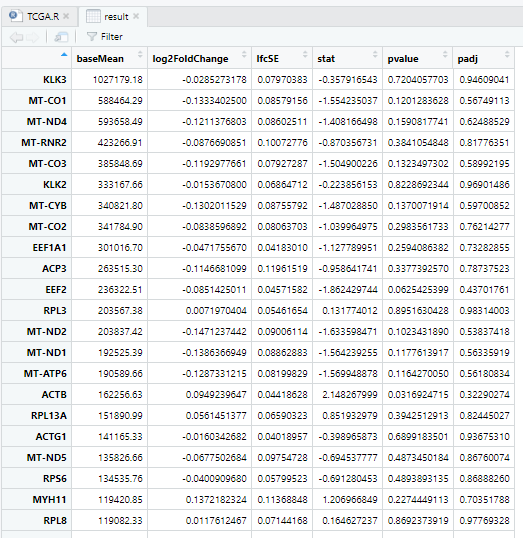
后面就可以进行下游分析了!