Amber是由多模块所组成的分子动力学软件,其工作流主要如下:
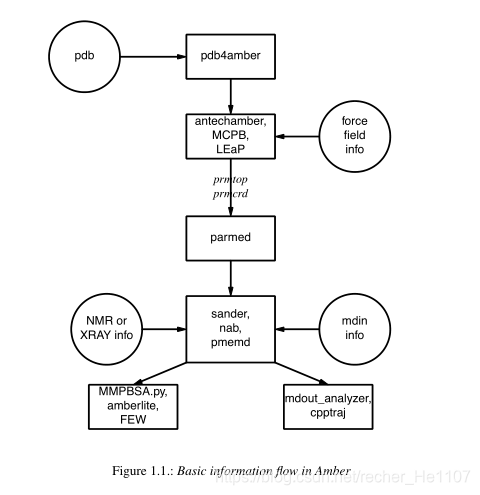
其中又分为:
1.系统预处理预处理(pdb预处理,LEAP,antechamber和gaff等)
2.运行动力学模拟(sander,pmemd等)
3.结果分析(mdout_analyzer.py,MMPBSA,cpptraj,FEW等)
1.系统预处理:
> 首先,进行分子动力学模拟我们需要对手中的pdb格式的3D蛋白质,核酸或者小分子配体,进行预先处理,使得其能够成为amber可识别可操作的形式,并解决其中的错误
一.蛋白质处理:
蛋白质主要存在的问题有
:
非必要结构: (1)非必须的水分子 (2) 配体
结构的不完整: (1)结构片段缺失(尤其loop);(2)残基结构不完整甚至出错;(3)二硫键问题.
很多软件多提供了自动处理蛋白结构的功能:如Schrodinger的Protein Preparation Wizard,MOE的Automated Structure Preparation等
.
我于此篇文章中写了如何用MOE处理蛋白
二.小分子处理:
小分子参数化的关键程序是Ambertools里的antechamber 会自动计算小分子的属性并获得参数,并处理小分子解决如下问题
Antechamber时, 你可能解决以下问题
:
1. 自动识别化学键以及原子类型
2. 判断原子等价性
3. 生成残基拓扑文件
4. 发现缺失的力场参数并提供合理的建议值
结合自带的GAFF 立场和parmchk程序 最终生成正确的LEAP可识别的 fcmod参数文件和mol2文件
详细的使用请参照此教程:
Amber基础教程B4:使用antechamber和GAFF模拟药物分子
三.读取处理好了的蛋白文件以生成amber可识别的 prmtop和inpcrd文件(坐标和轨迹文件)
涉及tleap的使用:
tleap #打开tleap source oldff/leaprc.ff03 #读取蛋白立场文件 mol =loadpdb protein.pdb #读取pdb文件 solvatebox mol TIP3PBOX 12.0 #为蛋白质添加水盒子(模拟体内的真实环境) charge mol #计算电荷 addions comp Cl- 7 # 分子模拟需要电中性,阳则补CL-例子,阴性则补Na+离子 aveamberparm mol mol.prmtop mol.inpcrd #保存蛋白的坐标文件和轨迹文件
三.读取小分子参数文件(fcmod):
tleap #打开tleap source leaprc.gaff #读取小分子立场文件 lig=loadmol2 ligand.mol2 #读取antechamber生成的小分子mol2文件 aveamberparm lig ligand.prmtop ligand.inpcrd #保存小分子的坐标文件和轨迹文件
上述的LEAP 都可以先写入tleap.in文件再执行
2运行分子动力学模拟:
首先需要的输入文件有
1. 最小化文件 min.in
2. 加热文件 heat.in
3. 平衡文件 density.in
4. 动力学文件 md.in
分别举行实例进行解释
> ##############################################
```bash
> 1.min.in
> minimise
&cntrl
imin=1, maxcyc=10000, ncyc=5000,
cut=10.0, ntb=1,
ntpr=500,
ntr=1, restraintmask=':1-199',
restraint_wt=10.0,
/
##############################################
> 2. heat.in
> heating
&cntrl
imin=0,irest=0,ntx=1,
nstlim=25000,dt=0.002,
ntc=2,ntf=2,
cut=10.0, ntb=1,
ntpr=500, ntwx=500,
ntt=3, gamma_ln=2.0,
tempi=0.0, temp0=300.0,
ntr=1, restraintmask=':1-500',
restraint_wt=1.0,
nmropt=1
/
##############################################
>3. density.in
>equilibration
&cntrl
imin=0, irest=1, ntx=5,
nstlim=250000, dt=0.002,
ntc=2, ntf=2,
cut=10.0, ntb=2, ntp=1, taup=2.0,
ntpr=500, ntwx=500, ntwr=5000,
ntt=3, gamma_ln=2.0,
temp0=300.0,
/
#############################################
>4. md.in
>10ns equilibration NPT
&cntrl
imin=0, irest=1, ntx=7,
ntb=2, pres0=1.0, ntp=1,
taup=1.0,
cut=10.0, ntr=0,
ntc=2, ntf=2,
tempi=310.0, temp0=310.0,
ntt=3, gamma_ln=1.0,
nstlim=5000000, dt=0.002,
ntpr=500, ntwx=500, ntwr=500,
/
############################################
**
imin =0 不执行最小化 =1 执行最小化
maxcyc=10000, ncyc=5000 最大循环数为10000 而前5000次循环采用最速下降法进行能量最小化
cut 其数值越大,越精准,单是会大大增加计算负担.不要使用低于8.0的值
ntb =1 周期性边界,恒容 =2 恒压 =0 不设定周期性边界
ntf = 1 进行力评估,计算出完整的相互作用 (默认值)
=2 ,忽略氢键相互作用
=3忽略所有键相互作用,
=4涉及H原子的角度且忽略所有键,
=5忽略所有键和角相互作用,
=6涉及H原子的二面体和所有键且忽略所有角度相互作用,
=7忽略所有键、角和二面体相互作用,
=8省略所有键、角、二面体和非键相互作用
ntr = 1标志用于使用谐波势限制特定原子
ntpr= 500 指定输出文件间隔,每500次输出一次结果至out文件中
restraintmask=':1-500' 限制残基长度
inhibitort_wt = 2.0 指定限制权重
irest =0 不进行重新启动,也就是输入坐标文件中的如果有坐标值将被忽略,时间步长计数将被设置为0。=1 重新启动.接着已有的文件进行分子模拟.
其参数有
有两种指令:
1.sander
2.pmemd
以实例进行展示
串行版本
sander -O -i md.in -p complex.prmtop -c density.rst -x md1.nc -o md.out -r md.rst -inf md.mdinfo
-O 文件若存在则重写文件
-i 指定输入文件
-p 指定输入的top文件(包含参数以及拓扑信息)
-c 指定输入的rst文件文件(包含坐标以速度信息)
-x 指定生成的mdcrd文件(轨迹文件)
-o 指定输出文件
-r 输出包含坐标和速度的重启文件(restrt)
-inf 输出包含模拟状态的MD信息文件
并行版本
mpirun -np 24 sander.MPI -O -i md.in -p complex.prmtop -c density.rst -x md1.nc -o md.out -r md.rst -inf md.mdinfo
-np 指定运行的核数
pmemd的是性能比sander更高的指令,其并行和穿行的用法和sander一致,但其独有gpu加速功能
pmemd.CUDA.MPI -O -i md.in -p complex.prmtop -c density.rst -x md1.nc -o md.out -r md.rst -inf md.mdinfo